
At last, 2016 has faded away into the past as the future of 2017 stands before us. For many, that change couldn’t have come soon enough. Just turn to Google for that proof.

For some though, the transition of a calendar means the inevitability of eroding profits. For those companies, each tick of the clock is a tick closer to the expiration of a patent. In the world of pharmaceuticals, that means a forthcoming generic drug that often provides a low cost alternative to an expensive name brand option. When patents are worth billions it’s worth keeping your eye on what’s to come.
What does a Pharmaceutical Patent do?
According to an article by news-medical.net, when a drug is under patent protection, only the company that holds the patent is allowed to manufacture, market the drug and eventually make profit from it. Patents in the industry typically last for 20 years from their application date, however, because patents are often applied long before a drug reaches the market, most drugs are only protected for between seven to twelve years once it goes on sale.
Why Does it Take so Long to Get a Drug from R&D into the Hands of a Patient?
The FDA publishes a tremendous amount of information on Investigational New Drug (IND) applications for new drugs. It’s a process that requires paperwork, approvals, exemptions and more. Currently, federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because sponsors will most likely be shipping investigational drugs to clinical investigators in many states, it must seek exemption from the requirement. Here’s an excerpt from the FDA’s website.
a new drug’s early preclinical development, the sponsor’s primary goal is to determine if the product is reasonably safe for initial use in humans, and if the compound exhibits pharmacological activity that justifies commercial development. When a product is identified as a viable candidate for further development, the sponsor then focuses on collecting the data and information necessary to establish that the product will not expose humans to unreasonable risks when used in limited, early-stage clinical studies.”
the drug is ready for human testing, the FDA steps in to begin its role and the process moves forward in one of any number of directions depending on the type of drug that’s being patented.
- IND | This drug is submitted by a physician who both initiates and conducts the investigation on the drug and under whose direction the drug would be administered or dispensed.
- Use IND | This is for use with an experimental drug in an emergency situation that does not allow time for submission of an IND in accordance with a number of guidelines. It is also used for patients who do not meet the criteria of an existing study protocol or if an approved study protocol does not exist.
- IND | This is used when an application is submitted for experimental drugs that show promise in clinical testing for serious or immediately life-threatening conditions while the final clinical work is conducted and the FDA review takes place.
It's important to note that there are two IND categories, Commercial and Research (non-commercial), and that they must contain information in three broad areas.
- Pharmacology and Toxicology Studies | Preclinical data must be included in order to permit an assessment as to whether the product is reasonably safe for human testing. Information on drug results on humans from foreign use is also relevant for this section.
- Information | Information pertaining to the composition, manufacturer, stability and controls used for manufacturing the drug substance and the drug product must be provided in the application. This information is assessed to ensure the company can meet the necessary supply needed for consumer demand.
- Clinical Protocols and Investigator Information | Detailed protocols for proposed clinical studies must have been considered for the FDA to assess whether the initial-phase trials will expose subjects to unnecessary risk. It must also include information on the qualifications of clinical investigators to assess whether they are qualified to fulfill their clinical duties. The application must show that a commitment will be made to obtaining informed consent from the research subjects, obtain review of the study by an instructional review board and adhere to IND regulations.
Once submitted, the sponsor must wait 30 calendar days before initiating any clinical trials. During this time the FDA can review the IND for safety to assure that those involved in human testing will not be subjected to unnecessary and unreasonable risk. Once this process is approved and eventually completed, the process moves from an IND application to a New Drug Application where the medicine is tested for safety and its overall effectiveness. Only 5% of medicines developed are able to complete this full process.
What Happens When Patents Expire?
As we discussed going into 2016, manufacturers desiring to sell the now-generic version of a drug that has gone off-patent do not need to prove the safety and efficacy of the drug since that has already been done. Instead, they submit a New Drug Application (ANDA) to the Food and Drug Administration (FDA) intended to demonstrate that the proposed generic is the same as the previously approved drug.
While it is commonly believed that the FDA’s drug approval process is slower than its foreign competitors, it is often faster and more willing to approve certain drugs. Seventy-five percent of the new drugs approved by both the FDA and European Medicines Agency (EMA) between 2006 and 2010 were first approved in the United States, while the FDA approved 32 of 35 prospective cancer drugs from 2003-2010. The EMA approved only 26.
You can read more about the overall investment that pharmaceutical companies spend to develop patents in last year’s blog.
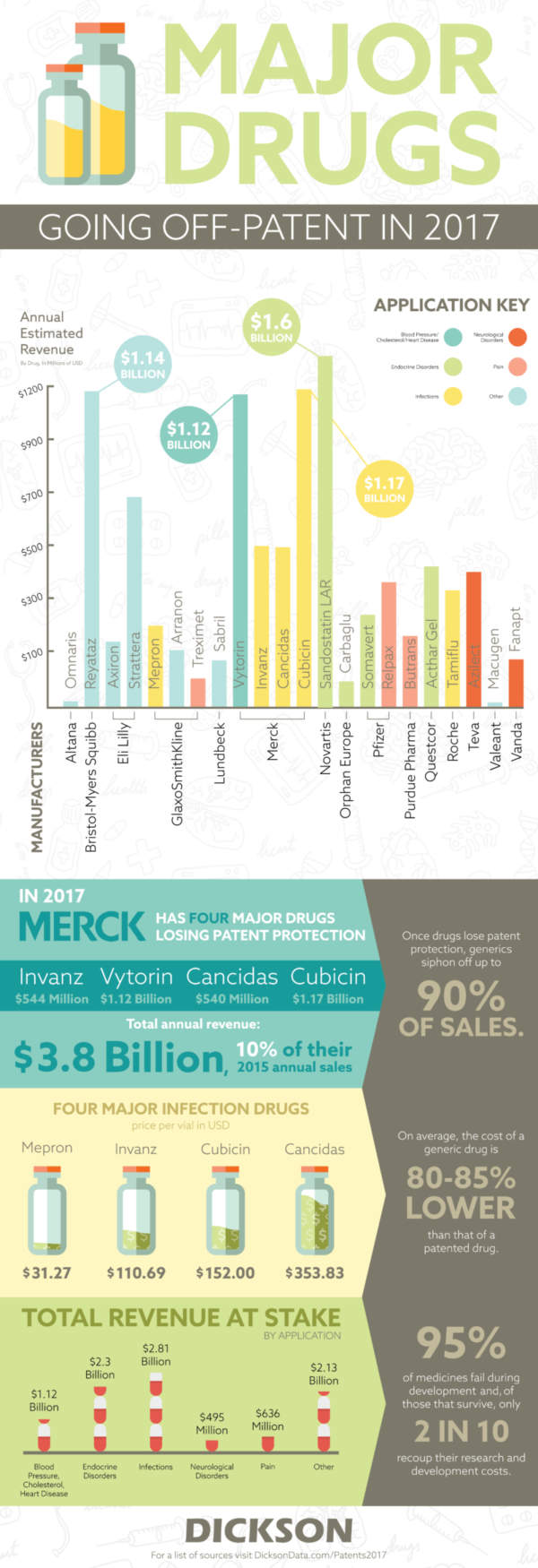
Who’s in line to lose the most?
Last year, it was Astra Zeneca. They lost patents on two major drugs, Cestor and Seroquel XR, that were worth a combined annual revenue of $7.34 billion. While the patent numbers aren’t nearly as large in the upcoming year, Merck still has the most to lose with four drugs losing protection. Their total value is estimated at nearly $3.4 billion. That accounts for nearly 10% of their sales based on their published 2015 financial data. While they won’t lose out on the entirety of the $3.4 billion, historical sales losses aren’t positive for pharma companies losing money to generics. When Pfizer lost its patent protection for Lipitor, which made $12.9 billion in sales at its highest point, it lost 90% of its revenue.
This often means large scale savings for patients in need of life-saving drugs. In 2008, there was often a saving of more than 70% on generics when compared to their brand named counterparts. While the major pharma brands are slated to lose out on revenue, the patient savings speak for themselves. It means that those in need of these drugs are as happy that 2016 is over as the rest of Google. There’s no reason to do a search on that.
Editor's Note: Patent law in the world of pharmaceuticals is complicated. Please visit the FDA for answers to a number of FAQ's on the topic.
Sources
- http://marketrealist.com/2016/03/patent-cliff-driver-generic-drugs-growth/
- https://newdrugapprovals.org/patent-expiry/
- http://www.pharmexec.com/what-happens-when-product-loses-its-patent
- http://www.drugstorenews.com/sites/drugstorenews.Com/files/GenericReport_2016.pdf
- https://www.drugpatentwatch.com/p/patent-expiry/2017
- http://www.centerlighthealthcare.org/images/uploads/Brand_Name_Drugs_with_Patent_Expirations_2013_-_2017.pdf
- http://sabril.net/
- http://files.shareholder.com/downloads/AMDA-GGC00/0x0x891323/375E37C4-25B2-4508-8E3F-22F0826018C5/Q1_Corporate_Release_UK.pdf
- http://www.xe.com/currencyconverter/convert/?From=DKK&To=USD
- http://www.pharmalive.com/glaxosmithkline-plc-2014/
- http://www.evaluategroup.com/Universal/View.aspx?type=Search&query=azilect
- https://www.pfizer.com/files/investors/presentations/q4performance_012913.pdf
- http://www.pfizer.com/system/files/presentation/2015_Pfizer_Financial_Report.pdf
- https://www.bms.com/ourcompany/Pages/keyfacts.aspx
- http://www.annualreports.com/HostedData/AnnualReportArchive/b/NYSE_BMY_2014.pdf
- http://s21.q4cdn.com/487185371/files/doc_downloads/2015-BMS-AR.pdf
- http://www.news-medical.net/health/Drug-Patents-and-Generics.aspx
- http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/NewDrugApplicationNDA/
- https://www.ucdmc.ucdavis.edu/clinicaltrials/IND/ind_documents/JournalofInvestigativeMedicineAugust2009.pdf
- http://www.slideshare.net/swati2084/ind-investigational-new-drug-application-and-nda-26468953
- http://blog.dicksondata.com/2015/12/drugs-losing-patent-protection/
- http://www.mercknewsroom.com/news-release/corporate-news/merck-announces-fourth-quarter-and-full-year-2015-financial-results
- http://www.forbes.com/forbes/welcome/?toURL=http://www.forbes.com/sites/johnlamattina/2016/04/14/u-s-drug-prices-to-decline/&refURL=&referrer=#4b561ae622c3
- http://healthsmart.com/SmarterHealth/GenericVsBrandDrugs.aspx
- https://www.goodrx.com/cancidas?drug-name=cancidas&form=vial&dosage=50mg&quantity=1&days_supply=&label_override=Cancidas
- https://www.goodrx.com/mepron
- https://www.goodrx.com/invanz?drug-name=invanz&form=vial&dosage=1g&quantity=1&days_supply=&label_override=Invanz
- http://www.nebraskamed.com/careers/education-programs/asp/restrictions/daptomycin
[/su_list]